 LAMMPS zostaÅ pierwotnie opracowany w ramach amerykaÅskiego Departamentu Energii LAMMPS zostaÅ pierwotnie opracowany w ramach amerykaÅskiego Departamentu Energii
W najbardziej ogÃģlnym sensie, LAMMPS integruje rÃģwnania ruchu Newtona dla zbiorÃģw atomÃģw, czÄ
steczek lub makroskopowych czÄ
stek, ktÃģre oddziaÅujÄ
za poÅrednictwem siÅ krÃģtkiego lub dalekiego zasiÄgu z wieloma rÃģÅžnymi warunkami poczÄ
tkowo-brzegowymi. Dla podniesienia wydajnoÅci obliczeniowej LAMMPS tworzy tzw. "listy sÄ
siadÃģw", dziÄki ktÃģrym ÅledziÄ moÅže ukÅad wzajemny czÄ
stek. Listy te sÅuÅžÄ
zoptymalizowaniu gÄstoÅci czÄ
stek na maÅych dystansach, tak Åže lokalna gÄstoÅÄ czÄ
stek nie jest nigdy zbyt duÅža. W maszynach rÃģwnolegÅych LAMMPS wykorzystuje techniki przestrzennego rozkÅadu czÄ
stek, aby symulacje podzieliÄ na mniejsze domeny 3D, z ktÃģrych kaÅžda jest przypisana do innego procesora. Procesory komunikujÄ
siÄ i przekazuj sobie jedynie informacje o czÄ
stkach sÄ
siednich subdomen leÅžÄ
cych na granicy domeny. LAMMPS jest najbardziej skuteczny (w sensie obliczeÅ rÃģwnolegÅych) dla systemÃģw, ktÃģrych czÄ
stki wypeÅniajÄ
prostokÄ
tne pudeÅko z grubsza o jednolitej gÄstoÅci.
Wszystkich zainteresowanych odsyÅam do bardzo obszernych materiaÅÃģw na stronie:
 Dodam jeszcze od siebie co waÅžne w systemach otwartych system posiada pre i post procesor, a wÅaÅciwie pre i post procesory, gdyÅž oprÃģcz oprogramowania przypisanego bezpoÅrednio do pakietu dystrybucyjnego moÅžna sobie jeszcze ÅciÄ
gnÄ
Ä wspomaganie, napisane w pytonie pod bardzo apetycznÄ
nazwÄ
Pizza.py. (http://pizza.sandia.gov/)
| 


 UÅžytkownicy Online
UÅžytkownicy Online









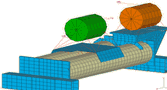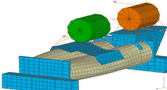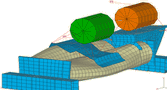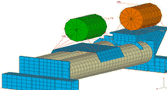

 ew thread" po prawej stronie.
ew thread" po prawej stronie.


